Lab Techniques for the DAT
Learn key DAT concepts related to gel electrophoresis, blotting, DNA sequencing, ELISA, cloning, and chromatography, plus practice questions and answers

learn everything you need to know about lab techniques for the dat
Table of Contents
Part 1: Introduction to biochemistry lab techniques
Part 2: Gel electrophoresis
a) Basic principles
b) Types
a. SDS-PAGE
b. Reducing SDS-PAGE
c. Native-PAGE
d. Isoelectric focusing
Part 3: Blotting methods
a) Basic principles
b) Types
a. Northern blot
b. Southern blot
c. Western blot
Part 4: DNA-based techniques
a) DNA sequencing via Sanger method
b) Polymerase chain reaction
a. RT-qPCR
b. cDNA library
Part 5: Enzyme-linked immunosorbent assay (ELISA)
a) Basic principles
b) Types
a. Direct
b. Indirect
c. Sandwich
Part 6: Molecular-biology techniques
a) Bacterial transformation
b) Molecular cloning
Part 7: Centrifugation and chromatography
a) Centrifugation
b) Gel filtration (size exclusion) chromatography
c) Ion-exchange chromatography
d) Affinity chromatography
Part 8: High-yield terms
Part 9: Questions and answers
----
Part 1: Introduction to biochemistry lab techniques
Welcome to our guide on experimental techniques in biology, tailored to assist you with the lab-focused content likely to appear on the DAT. This topic is high-yield, and comprehending these experimental methods will greatly benefit you during your exam. These techniques are often presented at a complex level, but don’t worry, as we aim to offer clear and concise explanations in this guide. Many find these techniques overwhelming, thinking they need to grasp every aspect, but we'll provide the essential information you need to excel!
While this guide is comprehensive, it serves as an invaluable resource for unraveling the intricate biology techniques featured on the DAT. We'll delve into a spectrum of techniques that you might encounter on your DAT, such as chromatography, molecular cloning, DNA sequencing, PCR, Blotting, ELISA, and gel electrophoresis. Our focus will be on the key elements pivotal for the DAT. Finally, we'll conclude with sample questions to gauge your proficiency.
----
Part 2: Gel electrophoresis
a) Basic principles
Gel electrophoresis segregates components within a mixture based on their size and charge. It is commonly applied to DNA, RNA, or various proteins. Imagine you have a mixture with five proteins and want to isolate one specific protein. Gel electrophoresis allows you to separate these proteins, so long as there is a difference in size or amino acid composition.
To separate the proteins, start by placing the mixture on a gel resembling a large sheet of Jell-O. Then, an electric field is applied across the gel with cathode (negative side) and anode (positive side). The molecules travel through the gel sheet in a sort of Jell-O swim meet.
These experiments leverage three critical properties: size, charge, and shape.
1. Size
Consider proteins with identical negative charges. When the electric field is activated, all proteins move towards the positive side but at different rates due to their sizes. Larger proteins travel more slowly through the Jell-O, akin to walking in waist-deep versus knee-deep water. Turning off the electric field halts protein movement.
2. Charge
Charge significantly influences migration speed. For proteins of similar size, higher net charges cause faster movement towards the positive end. Acidity and basicity of side chains determine a protein's net charge, with acidic side chains being negatively charged when deprotonated and basic side chains positively charged when protonated. These cumulative charges dictate movement speed and direction.
For nucleic acids like DNA and RNA, the phosphate groups between nucleosides bear a negative charge. This ensures a consistent negative charge across all DNA and RNA strands, known as an equal charge density. Consequently, charge uniformity removes its role in their migration speed, leaving only size and shape as influential factors.
3. Shape
Molecule shape influences migration speed. Streamlined or aerodynamic substances move faster through the gel. For example, consider a race car and a school bus with the same weight—the race car's aerodynamic design ensures that it moves faster than the school bus.
b) Types
a. SDS-PAGE
When it comes to separating proteins, the process is a bit more intricate than it is for DNA or RNA due to variable charge densities across proteins. While smaller DNA and RNA typically outpace larger strands, proteins might challenge this norm based on their distinct charge densities.
Consider this scenario: a large protein with numerous negative side chains versus a slightly smaller protein with fewer such side chains. Which will move faster—the larger one due to more negative charge or the smaller one because of its size? The proteins' 3D structures further complicate this, as varying shapes introduce aerodynamic considerations.
To circumvent the impact of diverse charge distributions and 3D shapes in protein separation, researchers employ SDS-PAGE. In simple terms, if your aim is to segregate proteins solely by size (amino acid count), SDS-PAGE is the go-to method!
In SDS-PAGE, researchers introduce sodium dodecyl sulfate (SDS) to their protein samples before gel electrophoresis. SDS denatures the protein and adds negative charges proportionate to the protein's size, creating an even charge distribution similar to DNA and RNA.
Visualize protein denaturation as unraveling a tangled knot. The primary structure is the string forming the knot, while secondary, tertiary, and quaternary structures are the entangled knot, and are essential for protein function in our analogy. SDS untangles this knot, leaving an unknotted string with negative charges based on its length. Shorter strings unfailingly outpace longer ones, relieving concerns about charge differences.

FIGURE 1: SDS-PAGE GEL
b. Reducing SDS-PAGE
In the realm of SDS-PAGE, there's a crucial detail yet to be uncovered: the addition of SDS completely unfolds proteins, excluding regions where disulfide bonds reside. Disulfide bonds form when two cysteine side chains within a protein link, similar to fastening two points along a string. This linkage can impede the protein's movement, slowing it down in gel electrophoresis.
To disrupt these disulfide bonds, a reducing agent comes into play that transforms the single disulfide S-S bond into two S-H bonds. This process is known as reducing SDS-PAGE. Employing this method ensures the elimination of higher protein structures, including any existing disulfide bonds and guarantees complete unfolding and standardizing the protein's behavior during separation.
c. Native-PAGE
At times, maintaining a protein in its natural form is crucial for certain studies. Once a protein is denatured, reverting it to its original shape—imagine re-tying a complex knot—is nearly impossible. If your research necessitates studying a protein in its native state, perhaps to observe its activity following the addition of an inhibitor, native-PAGE is the go-to method.
In native-PAGE, the process doesn't involve SDS or reducing agents, and the gel remains non-denaturing. This approach allows the protein to preserve its native shape, including its secondary, tertiary, and quaternary structures, facilitating research that requires the protein to maintain its original conformation.

FIGURE 2: NATIVE, SDS-PAGE, AND REDUCING SDS-PAGE GELS PROTEIN SHAPE
d. Isoelectric focusing
In the three electrophoresis methods we've covered, molecules typically migrate from the negatively charged side (cathode) to the positively charged side (anode). But, proteins might have a neutral or even positive net charge, making them move in the wrong direction!
Consider three proteins: Protein 1 has three negatively charged side chains (like two aspartates and one glutamate) and one positively charged side chain (e.g., lysine). Protein 2 is neutral, and Protein 3 has three positively charged side chains (like lysine, arginine, and histidine). Protein 1 has a net charge of -2, Protein 2 is at 0, and Protein 3 has a net charge of +3.
If you place all three proteins on the negative end of the gel, only the negatively charged Protein 1 moves towards the positive side. Proteins 2 and 3 remain unseparated. To address this, we employ isoelectric focusing.
Isoelectric focusing, like the earlier experiments, includes a stationary pH gradient within the gel, ranging from about 0 to 14. This gradient is alongside the electric field. The lowest pH sits near the gel's positive side (anode), while the highest pH is near the negative side (cathode). When molecules enter this gel, they move until they reach a region where the pH matches their isoelectric point (pI). The pI is where the protein holds a completely neutral charge.
How do we find a protein's isoelectric point? Consider the acidic and basic side chains. Acidic side chains are negatively charged when the pH is high but become protonated and neutral at a low pH (roughly 2). Conversely, basic side chains are positively charged at a low pH but become deprotonated and neutral at a high pH, approximately at 12. The isoelectric point is where the negative and positive charges balance, rendering the protein neutrally charged. At this pH, the protein halts its movement, unaffected by the poles' charges in the gel.

FIGURE 3: ISOELECTRIC FOCUSING GEL
----
Part 3: Blotting methods
a) Basic principles
Once you've successfully separated DNA, RNA, or proteins through gel electrophoresis, what follows next? Researchers often aim to pinpoint particular strands of DNA, RNA, or specific proteins. Blotting methods come into play here, aiding in the identification of these specific biomolecules. Three primary blotting methods—Northern, Southern, and Western Blots—are typically used. For each technique, it's crucial to grasp (1) the material under examination (DNA, RNA, or protein) and (2) the fundamental labeling technique used to pinpoint the target material.
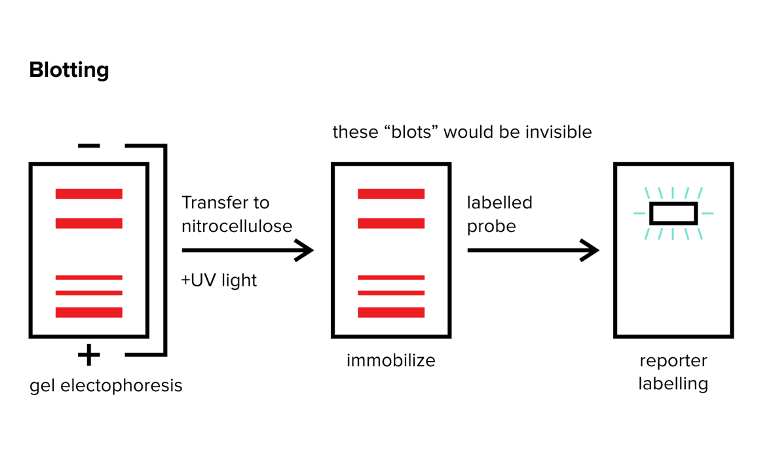
FIGURE 4: BLOTTING METHODS. THE RED BANDS ARE INVISIBLE AND MUST BE SEEN WITH A REPORTER
b) Types
a. Northern blot
You can employ a Northern blot to identify a specific RNA strand within your sample. After conducting gel electrophoresis to sort RNA strands by size, the next step involves transferring these RNA strands onto a different surface (usually nitrocellulose) and fixing them in place using UV light. Once the RNA is immobilized with UV light, you introduce a probe—a strand designed to specifically bind to the target RNA you're seeking. Usually, this probe is an RNA strand with a complementary sequence to the sought-after RNA, coupled with a label for visualization purposes.
To visualize where the probe has adhered, it's crucial to label the probe with a reporter. This reporter emits a signal that highlights where the probe has bound. This signal could manifest as colored light or a radioactive emission, among other forms.
For instance, imagine you've isolated RNA from various cancer cell types and want to detect if a particular transcription factor is being transcribed. If you know the processed mRNA sequence of this factor (e.g., 5’-AGCCAAU-3’), you can determine its presence in your gel. Immobilize the RNA onto nitrocellulose using UV light and introduce your labeled probe. Craft this probe to include a sequence complementary to the target RNA (e.g., 5’-AUUGGCU-3’: remembering the binding of the 3’ end to the 5’ end of the other) along with a reporter. Upon observing a signal from the reporter, you confirm the transcription factor's transcription within your gel.
b. Southern blot
A Southern blot mirrors a Northern blot but is focused on DNA rather than RNA. Following gel electrophoresis that separates DNA fragments, the DNA remains in a double-stranded state. To utilize a probe akin to the one in a Northern blot, the initial step involves denaturing the double-stranded DNA into single-stranded DNA. This step is crucial for enabling the binding of a probe with a complementary DNA sequence. The subsequent procedures closely resemble those used in a Northern blot.
c. Western blot
During a Western blot, the search for a specific protein involves a unique probe compared to its Northern and Southern counterparts. Unlike the nucleic acid-based complementary strands used in other blotting methods (which lack counterparts for proteins), Western blotting employs antibodies known for their high specificity in targeting individual proteins.
In this technique, two antibodies are typically utilized. The primary antibody is the first one deployed, binding specifically to the target protein. Unlike directly labeling this antibody with a reporter, a secondary antibody labeled with a light-emitting or radioactive substance is employed to recognize the primary antibody. Similar to the approach in Northern and Southern blots, the Western blotting process entails several stages. The first is gel electrophoresis, followed by protein immobilization and primary antibody application. Next, unbound primary antibodies are washed off, and secondary antibodies are applied. Again, unbound antibodies are removed. Following this process, researchers can look for the signal.

Gain instant access to the most digestible and comprehensive DAT content resources available. Subscribe today to lock in the current investments, which will be increasing in the future for new subscribers.